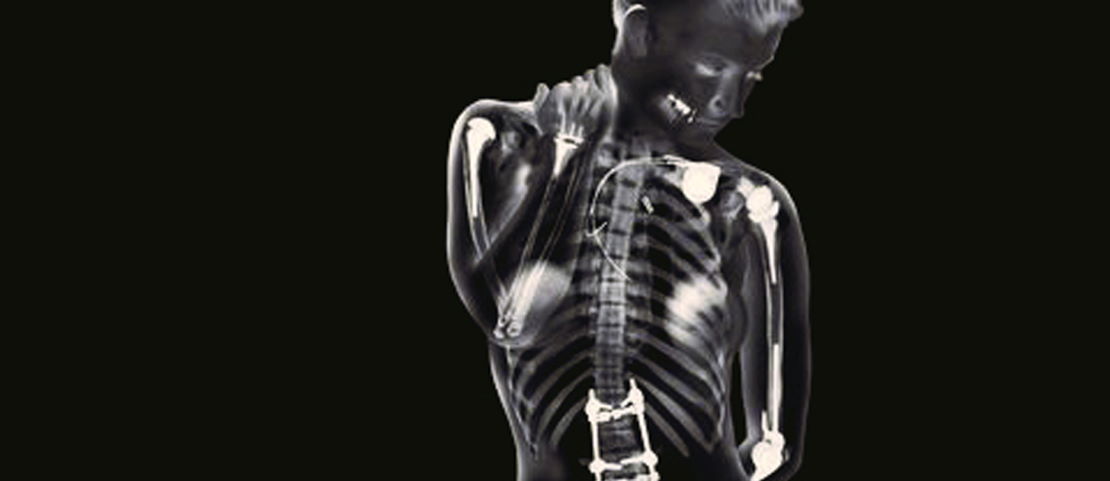

Implanted medical devices covers everything from breast implants to pacemakers to contraceptives to vaginal mesh to artificial hips. The list goes on but basically includes anything that’s put inside you and stays there.
In the UK, regulators have received 62,000 of these reports in the years between 2015 and 2018, a third of which caused serious issues for the patient and over 1,000 of which resulted in death. In figures from the US, the figures paint an even starker picture. Over the last 10 years, the FDA has collected 5.4m reports of the same nature. This includes 1.7m injuries and over 80,000 deaths. Almost half a million of these mentioned an explant, meaning surgery was required to remove the device.
So, what’s gone wrong?
Regulations Aren’t Regulating
Firstly, a caveat. I think it’s important to say that whilst we’re looking at some bleak statistics, the benefits that the invention and adoption of many implanted devices have been off the charts. A successful medical device, just like a new ‘wonder drug’ can change thousands or millions of lives almost instantly which, counter intuitively, could be part of the problem.
When a new device hits the market that does something new or more effectively than its predecessor, the demand can be astronomical as patients and hospitals all clamour to be the first to use it. Doctors can forge ties with manufacturers to ensure that they, and their clinic or practice, is the first to bring it to market.
This means that once claims are made by a manufacturer as to a device’s benefits, there are massive pressures placed on the regulators to help get this to market, both from physicians and patients.
Firstly, let’s look at how it works in Europe. In the European Economic Area (the EU plus Iceland, Liechtenstein and Norway), medical devices need to be given a CE mark in order to be brought to market, the same mark that is given to all products deemed fit for sale in the region.
There are 58 bodies that can award a CE mark in Europe and, crucially, if one says no then a company looking to get their product approved can go to another to attempt to gain approval there. That’s the same whether you’re bringing a breast implant, games console or new spoon to market in the EEA. Then it’s up to the individual territories to approve medical devices on their own terms.
One pronounced example of this system being problematic, as reported by the BBC, was with the Nanostim pacemaker. At the time marketed as the first leadless pacemaker to sit inside the heart, many patients were delighted to be fitted with the device.

However, what those patients didn’t know is that the device was turned down by safety bodies in Germany due to a lack of evidence from clinical trials of its success, whereas it was approved in the UK by the British Standards Institution.
Ultimately, the Nanostim was identified as the cause for two deaths and over 90 serious injuries to patients as issues arose like the devices failing or parts falling off. Perhaps if the German safety body which turned down the device for use were able to share their findings or concerns, these could have been avoided.
In the US, it’s a different process. Approvals there are controlled by the Food & Drug Administration (FDA) but the pressure to bring new devices to market quickly remains the same. One avenue open to companies looking for faster approvals is an expedited process based on a device’s similarity to a predecessor.
If a device is deemed to be ‘substantially equivalent’ to a device that comes from the same ‘family’ that came before it (since 1976 when the regulation was introduced) the approval process can be sped up. This means that new devices could be gaining FDA approval and being brought to market based on their similarity to 20, 30 or 40-year-old products.
But it’s important to keep sight of the truth and motivations. Patients want these devices because they want to make their lives better. This is understandable. Companies want to bring these devices to market to help patients, while capitalising financially in the process. This means that patents, technologies and results of trials are fiercely protected by companies who don’t want vital information about their products being leaked.
What Can be Done?
Further to the last point, many organisations and governments are calling for more transparency. Whilst many have been aware of individual incidents occurring, the revelations this week have revealed the scale of the problem and led to calls for new regulations and measures to be put in place to prevent, or at least identify and solve, widespread problems with implanted devices.
One call that has been made by the Royal College of Surgeons in the UK and echoed by various bodies in the US has been for a register of devices. This would mean that every device inserted into every patient is logged in their medical records which means that, should a defect be identified, this can be flagged for other patients who have had the same devices inserted.
In the US this system already exists, as Unique Device Identifiers (UDIs) have been standard practice in some devices for a number of years. The issue is that there has not, up to now, been a place to include this information on patient records, meaning that whilst the numbers do exist, they aren’t being tracked or recorded.
Another method of incident prevention suggested has been deterrence, through making it easier for patients to make claims if they have been subjected to injury as a result of an implanted device. This already exists in Scandinavia, which is where it’s known as the Nordic Scheme.
First rolled out in Sweden in 1975, the scheme ensures easy access for patients to compensation if their injury is deemed avoidable. The compensation is paid for by the device manufacturers themselves which avoids costly, lengthy and sometimes traumatic court battles. However, critics argue that this system is also a quick fix for the manufacturers themselves and that settlements are often significantly lower than those given out in clinical negligence trials.
Whilst making it easier to claim could be a deterrent and make medical companies pursue more stringent trials of their products, it’s surely reports like these that will really change the landscape of the market. But only time will tell.
Where Do We Go From Here?
There’s already been a big fallout from the reports and calls for tighter regulations are already increasing in their frequency and volume. In the US, the FDA has already made UDI numbers standard across all devices (scheduled to happen before these reports broke) and new regulations are being introduced so that devices can only be deemed ‘substantially equivalent’ to devices that are 10 years old or younger.
New, stricter, EU legislation is also being introduced in 2020, but some critics are now saying that doesn’t go far enough.
What we are left with is a situation in which governments are blaming companies for trying to get around regulations and companies are pointing the finger back, saying that the regulations need to be tougher. That’s all very murky but what is for sure is that patients are suffering, which is why the new legislations are going to be welcomed by many.
One certainty we are left with is that the general public is now more aware than ever of this issue, which can only be a good thing. My only hope is that the same public isn’t demonising an industry which has provided massive benefits and saved, or enhanced, millions of lives.

In this episode I talk to ex-physician gone start-up CEO, Avi Fischer, about the future of Catheter Ablations and how EP Frontiers is disrupting the industry. Click to listen.
.jpg)
With the limited data we have so far, PFA has proven to be safer than other ablation energies and to provide quicker recovery times. But is it ready to revolutionise cardiac ablations? To find out, I spoke to five leaders from start-ups in the space.

In this episode, I'm joined by Steven Haken and Deborah Rizzi from market access and reimbursement specialists Odelle Technology to discuss how to take a medical device to market.

I caught up with the Head of Digital Health, Diagnostics & Monitoring at Biotronik, Ken Nelson, to talk about the increasing value of data within the space.
Comments.